hongkongdoll 免费视频 芳醇型偶氮污辱物紫外光谱量子力学指认及不同共轭态偶氮键电子引发谈论

偶氮污辱物(Azo Contaminants, ACs)是以氮氮双键为中枢结构的有机分子, 是常见的化学试剂、化工中间体和末端家具, 在pH检测、非线性光学材料合成(Samanta et al., 2018)、活性氧摆脱基(Reactive Oxygen Species, ROS)顽强及日用品染色等方面有横暴哄骗.偶氮污辱物对生态系统具有不良影响, 其次坐蓐物—归附性苯胺类化合物更是具有“三致”作用.因此, 联系偶氮污辱物的处理处置成为较受关注的谈论地点(Petrella et al., 2013).常见的设施有高档氧化、光催化、吸附、生态建筑等(Yang et al., 2014; Zhao et al., 2019).现时, 东说念主们对偶氮污辱物的光降解谈论相同稳重于盘考偶氮化合物与功能材料的相互作用(Petrella et al., 2013; Yang et al., 2014; Cai et al., 2017; Guan et al., 2019; Zhao et al.hongkongdoll 免费视频, 2019), 针对其自己与光子的相互作用鲜被关注, 然则这既是光反应的核神思理之一, 亦然污辱物浓度定性定量分析的表面基础.因此, 谈论“光子-偶氮污辱物结构-电子引发”间的耦合模式不错填补空缺, 为环境监测、环境建筑、环境功能材料开垦及非线性光学材料合成提供表面依据.Gagliardi等(2004)通过表面计算发现了偶氮苯分子光致异构经过中粗劣单线态的存在, 从而解释了偶氮苯光化学的波长依赖征象, 为光催化波长、光源及介质的采纳提供了依据.Döbbelin等(2016)通过对偶氮苯分子结构的调控, 在365 nm光照下到手剥离石墨晶体得到了单层石墨烯, 在环境友好的非强酸强碱要求下竣事了石墨烯制备.此类谈论为光照环境中污辱物转移革新、碳基环境材料的开垦设立了讲究的表面基础.
偶氮污辱物分子与光子相互作用的具象化阐扬为紫外-可见领受光谱(下称紫外光谱), 在一定浓度领域内, 吸光度与浓度具有讲究的线性关系.实验紫外光谱具有较长的谈论历史, 是定性定量、监控反应经过的有劲器用.溶剂环境中, 一般分辨率的紫外光谱相同被以为清寒细腻领略能力, 溶剂拉平效应、氢键或“溶质-溶剂”络合物的酿成迁延了细腻结构.已有谈论使用多高斯峰拟合(Zhang et al., 2012)、差分光谱(Yan et al., 2016)等本事对分子构象或复杂结构宦官能团散布进行探索, 但离细腻结构领略仍有差距.计算化学不错通过振子强度与引发能谈论领受峰强度与位置, 是有劲的光谱指认器用.相同以为偶氮分子中n电子跃迁至π反键轨说念(n→π*)是最容易发生的跃迁经过(引发能大致为3.00 eV), 但此经过引发电子(n电子)开始于2个氮原子, 其占据通盘分子的摩尔比例为8.33%(以偶氮苯为例), 此局域引发经过与实验光谱主要领受峰设立关联需要更明确的实验或表面计算把柄.Daniel等(2011)在PBE0/TZVP计算水平下谈论了6种偶氮染料的紫外领受光谱, 指出n→π*跃迁尽管是最容易发生的跃迁, 但其领受(振子)强度为0(跃迁禁阻), 因此不合实验光谱第一权臣领受峰作念孝敬, 许多表面谈论齐抓有类似不雅点(Manzoni et al., 2019).但若因此全齐否定偶氮键对紫外领受的孝敬, 亦清寒明确的科学把柄.
香蕉视频在线观看一直看一直爽基于此, 本谈论以偶氮苯和甲基橙为方针污辱物, 通过对比“顺反式”偶氮苯分子的实验与计算光谱, 谈论不同共轭景色下偶氮键的紫外领受特点, 汇报“偶氮键”对“偶氮污辱物”领受光谱的孝敬情况, 谈论弱相互作用对领受光谱的影响.从分子轨说念、振子强度等角度, 探明领受光谱与电子散布之间的关系, 揭示显色行径内在机理, 为偶氮污辱物紫外光谱指认、污辱物光催化、污辱物环境行径及分子开关忖度打算等谈论提供表面依据.
2 材料与设施(Material and methods) 2.1 实验设施谈论使用的两种偶氮污辱物分别为偶氮苯(CAS No.103-33-3, 上海阿拉丁)和甲基橙(CAS No.547-58-0, 上海阿拉丁), 除酒精(上海国药)为色谱纯外, 其他试剂均为分析纯(上海国药).实验紫外光谱测量波长为200~600 nm(上海元析UV8000-S, 10 mm石英比色皿), 使用0.1 mol·L-1 HCl和NaOH诊治被测溶液pH到2、7和12, 污辱物浓度为10 μmol·L-1, 溶剂为1%酒精水溶液(助溶), 实验用水电阻率为18.2 MΩ·cm-1, 总共实验均在(10±2) ℃下进行.为了便于实验室间对比, 后续补充了(22±2) ℃下的实验光谱, 与10 ℃下的实验光谱比较, 其峰偏移小于0.1%, 不存在权臣区别.
2.2 表面计算设施本文的计算责任东要包括以下部分: ①基态分子构型的优化和振动分析, 以得到安祥结构;②以①为基础的表面紫外光谱指认(使用含时密度泛函设施);③分子能源学模拟, 设立团簇模子, 接洽紫外领受光谱.其中, ①和②使用高斯09w计算(Frisch et al., 2009), ③的分子能源学部分使用Gromacs2018.8模拟(Abraham et al., 2015).波函数分析使用Multiwfn进行(Lu et al., 2012).除非额外声明, ①和②均在SMD模子的水溶液环境中进行计算并使用GD3(BJ)设施进行色散矫正(Marenich et al., 2009; Grimme et al., 2011), 以知足弱相互作用的计算需求.鉴于M06-2x和PBE38分别在结构优化和大共轭分子引发态谈论中的讲究阐扬(Goerigk et al., 2010; Walker et al., 2013), ①中使用M06-2x/def2-tzvp进行优化和振动分析以得到局部最安祥的分子构型, ②中使用PBE38/def2-tzvpp进行含时密度泛函(Time-Dependent Density Functional Theory, TDDFT)计算以得到表面紫外领受光谱, 为尽可能粉饰感敬爱敬爱的光谱领域, 计算引发态数陶冶为100.
分子能源学模拟在10 nm边长的周期性立方体水盒子中进行, 水分子使用SPC/E模子, 水分子和污辱物分子模拟参数分别由琥珀14SB力场(Amber14SB)和通用琥珀力场(General amber force field, GAFF)界说(Maier et al., 2015, Wang et al., 2004), 原子电荷为浪漫性静电势拟合电荷(0.5)(Restrained Electrostatic Potential, RESP2(0.5))(Schauperl et al., 2020).动手结构使用Packmol 18.169搭建(Martinez et al., 2009).
3 后果与盘考(Results and discussion) 3.1 不同共轭环境中偶氮键电子跃迁特点偶氮苯(Azobenzene, AB)是偶氮染料分子的中枢结构hongkongdoll 免费视频, 具有顺式与反式两种构型(图 1A、1B), 含有不同共轭景色下的偶氮键(顺式→局部共轭, 反式→全局共轭).顺式能量高于反式, 在特定波长明朗“引发”下, 反式不错革新为顺式, 撤去引发祥后, 顺式通过热弛豫回到反式, 因此, 反式是环境中的主要构型(Gagliardi et al., 2004; Duarte et al., 2014).位阻效应、色散力及异构化能垒(Cattaneo et al., 1999; Duarte et al., 2014; Titov et al., 2015)共同导致顺式偶氮苯能在较高能级相对安祥存在.顺式和反式具有类似的轨说念散布特征, 即两者的最高占据轨说念(Highest Occupied Molecular Orbital, HOMO)和最低未占据轨说念(Lowest Unoccupied Molecular Orbital, LUMO)齐对应各自的48、49号分子轨说念.前方分子轨说念体现了反式偶氮苯的举座芳醇性(图 1a~1d), 其HOMO轨说念体现了苯环与偶氮键结构中沿分子平靠近称散布的电子结构(图 1c), 其HOMO-1轨说念体现出偶氮键隔邻σ键和孤对电子(图 1d), 其反键轨说念亦具有权臣的芳醇特征(图 1a、1b).
 图 1
顺反式偶氮苯结构与前方分子轨说念等值面图
(0.05 a.u.)(白(蓝)椭球体分别代表正(负)相位, 括号中数字为分子轨说念序号)
Fig. 1
Cis-trans azobenzene structure and Iso-value surface diagram of its frontier molecular orbital
(0.05 a.u.)(white (blue) ellipsoid represents positive (negative) phase, respectively. Numbers in brackets are the molecular orbital number)
图 1
顺反式偶氮苯结构与前方分子轨说念等值面图
(0.05 a.u.)(白(蓝)椭球体分别代表正(负)相位, 括号中数字为分子轨说念序号)
Fig. 1
Cis-trans azobenzene structure and Iso-value surface diagram of its frontier molecular orbital
(0.05 a.u.)(white (blue) ellipsoid represents positive (negative) phase, respectively. Numbers in brackets are the molecular orbital number)
顺式偶氮苯HOMO轨说念中不错不雅察到多种电子结构的羼杂征象(图 1g), 苯环上具有较强的芳醇性特征, 在偶氮键上能够不雅察到n电子结构(图 1g中偶氮键上方两个等值面椭球体), 而在辘集苯环与偶氮键的C—N键上则不雅察到π-σ的羼杂结构特征(图 1g中苯环平面两侧的等值面椭球体和毗邻C—N键上沿轴向散布的等值面椭球体).举座而言, 顺式结构的波函数构造显得愈加“凌乱”和“落空”.由此可见, 顺式偶氮苯的举座芳醇性弱于反式偶氮苯, 偶氮键中的n电子(或π电子)在顺式结构中可能不参与全局共轭.换言之, 偶氮键在反式偶氮苯中处于一个“大”共轭体系中, 顺式则不是.为了进一步考证偶氮键的共轭环境, 针对性地进修π电子散布情况, 引入局部轨说念定位函数(Localized Orbital Locator, LOLπ)(Schmider et al., 2000; Lu et al., 2020), LOL是基于电子动能密度(Kinetic Energy Density, KED)的意见密度泛函函数, 能清亮直不雅地展现体系中的化学键区域(图 2b、2d).在两种结构中, π电子密度散布特征类似, 均可粗劣分辩为两部分: 苯环π电子及偶氮键π电子(图 2a、2c), 但无法判断它们之间的调换(共轭)情况.LOLπ函数等值面标明, 反式偶氮苯中总共π电子酿成了一个完好的“大”共轭结构(图 2b), 体现出偶氮键桥接效应.顺式偶氮苯中的π电子则酿成3个相对孤独的部分(图 2d), 因此, 两者引发特点必有各异.
 图 2
偶氮苯π电子密度等值面图(0.05 a.u.)(a, c)与LOLpi函数等值面图(0.35 a.u.) (b, d)
Fig. 2
Isosurface of azobenzene π electron density iso-value surface(0.05 a.u.)(a, c) and LOLpi function iso-value surface (0.35 a.u.)(b, d)
图 2
偶氮苯π电子密度等值面图(0.05 a.u.)(a, c)与LOLpi函数等值面图(0.35 a.u.) (b, d)
Fig. 2
Isosurface of azobenzene π electron density iso-value surface(0.05 a.u.)(a, c) and LOLpi function iso-value surface (0.35 a.u.)(b, d)
鉴于PBE38(HF因素37.5%)表面设施在共轭体系引发态谈论中的讲究阐扬(Goerigk et al., 2010; Liu et al., 2020a; 2020b), 本谈论使用此表面设施对两种偶氮苯分子进行TDDFT计算, 得到的表面计算光谱与实验光谱高度重合, 为后续的指认责任奠定了讲究基础(图 3a).全局芳醇性使得π电子能在通盘分子中相对摆脱地转移, 促使引发能镌汰、领受峰红移.引入反式偶氮烯分子( 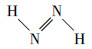 )用于对比谈论, 它是由偶氮键和2个氢原子构成的浅薄分子, 能反馈隧说念偶氮键的光谱特点(Tang et al., 1994).反式偶氮烯的共轭领域被浪漫在偶氮键且氢原子唯有一个s电子(1S1), 其共轭领域小于反式或顺式偶氮苯(顺式偶氮苯在苯环指引下, 亦有强于偶氮烯而弱于反式偶氮苯的电子离域情况).计算后果标明, 3个分子中反式偶氮烯的第一引发能(E1)最高(表 1), 达3.216 eV, 但振子强度为0, 属于跃迁禁阻.反式偶氮烯的实验光谱在355 nm处却有很强的领受峰(Tang et al., 1994), 可能因为跃迁磁偶极矩孝敬导致(表 1);其次, 偶氮烯与水分子过头他阳离子相互作用导致振子强度加多或其他跃迁领受峰红移(此实验光谱在高浓度(0.21 mol·L-1)乙酸钠缓冲溶液中测定(Tang et al., 1994)).
)用于对比谈论, 它是由偶氮键和2个氢原子构成的浅薄分子, 能反馈隧说念偶氮键的光谱特点(Tang et al., 1994).反式偶氮烯的共轭领域被浪漫在偶氮键且氢原子唯有一个s电子(1S1), 其共轭领域小于反式或顺式偶氮苯(顺式偶氮苯在苯环指引下, 亦有强于偶氮烯而弱于反式偶氮苯的电子离域情况).计算后果标明, 3个分子中反式偶氮烯的第一引发能(E1)最高(表 1), 达3.216 eV, 但振子强度为0, 属于跃迁禁阻.反式偶氮烯的实验光谱在355 nm处却有很强的领受峰(Tang et al., 1994), 可能因为跃迁磁偶极矩孝敬导致(表 1);其次, 偶氮烯与水分子过头他阳离子相互作用导致振子强度加多或其他跃迁领受峰红移(此实验光谱在高浓度(0.21 mol·L-1)乙酸钠缓冲溶液中测定(Tang et al., 1994)).
 图 3
偶氮苯实验与计算紫外-可见领受光谱
(a.不同pH要求下反式偶氮苯分子实验和计算光谱, b.顺(反)偶氮苯计算光谱, exp.为实验光谱, theo.为计算光谱)
Fig. 3
Experimental and theoretical UV-Vis spectrum of azobenzene
(a.Experimental and theoretical spectrum of trans-azobenzene under different pH conditions, b.Theoretical spectrum of cis (trans) azobenzene)
表 1 偶氮烯和睦反偶氮苯引发态关系指数(计算得到)
Table 1 Corresponding index of the excited states of diazene and cis-trans azobenzene (calculated)
图 3
偶氮苯实验与计算紫外-可见领受光谱
(a.不同pH要求下反式偶氮苯分子实验和计算光谱, b.顺(反)偶氮苯计算光谱, exp.为实验光谱, theo.为计算光谱)
Fig. 3
Experimental and theoretical UV-Vis spectrum of azobenzene
(a.Experimental and theoretical spectrum of trans-azobenzene under different pH conditions, b.Theoretical spectrum of cis (trans) azobenzene)
表 1 偶氮烯和睦反偶氮苯引发态关系指数(计算得到)
Table 1 Corresponding index of the excited states of diazene and cis-trans azobenzene (calculated)
水环境中反式偶氮苯与顺式偶氮苯的E1分别为2.775 eV(447 nm)和2.897 eV (428 nm), 证据π电子的全局共轭有意于S0→S1引发能的镌汰, 而较小的振子强度标明反式偶氮苯实验光谱中320 nm处的最大领受峰(图 3a)由其他跃迁经过孝敬.反式偶氮苯的计算光谱中未能不雅察到相应的447 nm的强领受峰(图 3b黑线), 然则其实验光谱在422 nm位置不雅察到一个弱的展宽较大的领受峰(图 3a中422 nm绿框隔邻), 此处领受存在多种可能性: 有谈论以为此处领受是偶氮苯质子化导致(童沈阳, 1980), 也有可能是跃迁磁偶极矩孝敬(0.974 a.u.), 同期不摒除顺式偶氮苯的存在, 偶氮苯顺反式异构化能约为1.09 eV (25.136 kcal·mol-1)(Yu et al., 2015), 系统中有可能存在一丝顺式偶氮苯, 实验光谱较深紫外区的领受峰亦证据了这一丝(图 3a中红框229 nm处领受峰), 与顺式偶氮苯计算光谱高度一致(图 3b中230 nm处), pH=12时229 nm处高斯峰形破缺由高浓度Na+导致, 因为pH=12对应浓度的Na+(10 mmol·L-1 NaOH)在 < 210 nm波万古存在权臣紫外领受, 与污辱物的峰肖似后导致本来对称的峰形失去了对称性.
分子引发经过是电子转移经过, 将引发前电子位置界说为“空穴”, 将引发后电子位置界说为“电子”(Liu et al., 2020c), 就不错直不雅地进修电子转移.环境化学中顺心波长领域一般为200~600 nm, 对应的顺式偶氮苯跃迁经过包括S0→S1到S0→S10.在这10个跃迁经过中, 最低对应S0→S1(428 nm, f=0.031), 振子强度大于0.1的跃迁经过包括S0→S2(247 nm)与S0→S6(231 nm)(表 1), 是最需要关注的3个跃迁.与之对应的, 反式偶氮苯应关注的引发态为S1、S2及S6(表 1).图 4中清亮地展现了顺式偶氮苯3个引发经过中的电子变化.参与S0→S1跃迁的电子在被“引发”之前主要散布在偶氮键沿C—N键位置, 由N原子及C—N键成键电子构成(图 4a), “引发”后的电子呈现出罕见典型的π散布特征, 应将其指以为n→π*引发.关于“电子”而言, 此计算级别下LUMO轨说念的孝敬率高出了94%;关于“空穴”而言, 主要孝敬轨说念为HOMO轨说念(48号分子轨说念), 孝敬率为78%.因此, 顺式偶氮苯的S0→S1跃迁主要对应其HOMO-LUMO轨说念跃迁;S0→S2和S0→S6是能量更高的引发经过, 从空穴-电子等值面进修基本不错指以为π→π*引发(图 4b、4c), 但仍然包括部分n→π*因素.总而言之, 在非全局共轭体系中, 偶氮键第一引发态为n→π*, 但振子强度较弱(f=0.031), 标明“一身”偶氮键对顺式结构中的紫外领受孝敬有限, 即实验光谱中测得的第一个主要领受峰不是由S0→S1孝敬, 不成指以为n→π*跃迁类型.
 图 4
顺式偶氮苯S0→S1、S0→S2、S0→S6引发时电子散布变化
(MO是分子轨说念Molecular Orbital的缩写, MO之后的数字代表轨说念序号;a、b、c分别对应第1、2、6引发态;空穴和电子等值面图
(0.002 a.u.)中白色和红色椭球体分别代表“电子”和“空穴”;分子轨说念等值面图(0.05 a.u.)中白色和红色椭球体分别对应正负相位)
Fig. 4
The electron distribution of cis azobenzene during the excitation of S0→S1, S0→S2, S0→S6
(MO is the abbreviation of Molecular Orbital, and the number after MO represents the orbital number. In the "electron-hole" column, the white and red ellipsoids represent the "holes" and the "electron" isosurface
(0.002a.u.) respectively. In the "MO" column, the white and red ellipsoids represent the positive and negative phases of the wave function (0.05 a.u.), respectively)
图 4
顺式偶氮苯S0→S1、S0→S2、S0→S6引发时电子散布变化
(MO是分子轨说念Molecular Orbital的缩写, MO之后的数字代表轨说念序号;a、b、c分别对应第1、2、6引发态;空穴和电子等值面图
(0.002 a.u.)中白色和红色椭球体分别代表“电子”和“空穴”;分子轨说念等值面图(0.05 a.u.)中白色和红色椭球体分别对应正负相位)
Fig. 4
The electron distribution of cis azobenzene during the excitation of S0→S1, S0→S2, S0→S6
(MO is the abbreviation of Molecular Orbital, and the number after MO represents the orbital number. In the "electron-hole" column, the white and red ellipsoids represent the "holes" and the "electron" isosurface
(0.002a.u.) respectively. In the "MO" column, the white and red ellipsoids represent the positive and negative phases of the wave function (0.05 a.u.), respectively)
反式偶氮苯是环境中的主要构型, 谈论其实验和计算光谱对偶氮类污辱物检测和光催化波长采纳具有率领真谛.由图 3a可知, 计算光谱很好地吻合了实验光谱, 其波长缺陷仅为6 nm.反式偶氮苯的两个苯环具有很强的共面性(二面角179.99°), 之前也证据了其举座芳醇性(图 1b、1c和图 2a、2b). 推测偶氮苯分子的第一个“权臣”领受峰(图 3a实验光谱320 nm处峰)势必有π电子引发参与.“空穴-电子”分析标明, 反式偶氮苯的S0→S1引发主要由偶氮键参与, 具有权臣的n电子和σ电子特征, 引发能为2.775 eV, 中心峰位置在447 nm处, 且为跃迁禁阻(f=0), 其“空穴”和“电子”具有权臣的π对称性(图 5a).反式偶氮苯的S0→S2跃迁是事实上的HOMO→LUMO跃迁, 分别对应第48号到49号分子轨说念跃迁(顺式的S0→S1跃迁对应HOMO→LUMO跃迁), 其引发能为3.917 eV, 中心峰位于316.57 nm处, 与实测峰位置(320 nm)偏差小于5%.反式偶氮苯的S0→S2引发有许多“大共轭”结构的π电子参与, 引发轨说念构成相对单纯, “空穴”和“电子”主要由48号和49号轨说念孝敬(图 5b).48号轨说念波函数等值面图标明苯环和偶氮键上的π电子均对S0→S2引发作念出了弥留孝敬, 且引发后电子仍呈较强的π对称性(图 5b).由此可见, 反式偶氮苯的实验第一领受峰应当指以为π→π*跃迁, 且主要官能团—苯环和偶氮键齐有孝敬, 而非唯有后者孝敬;与之相背, 偶氮键引发经过具有很强的局域特征, 领受强度很弱, 实验光谱未能体现.由此可见, 偶氮键在电子引发经过中施展了“桥接作用”, 能够促进反式偶氮苯领受峰红移.
 图 5
反式偶氮苯S0→S1、S0→S2、S0→S6引发时电子散布变化
(MO是分子轨说念molecular orbital的缩写, MO之后的数字代表轨说念序号;a、b、c分别对应第1、2、6引发态;空穴和电子等值面图
(0.002 a.u.)中白色和绿色椭球体分别代表“电子”和“空穴”;分子轨说念等值面图(0.05 a.u.)中白色和绿色椭球体分别对应正负相位)
Fig. 5
The electron distribution of trans-azobenzene during the excitation of S0→S1, S0→S2, S0→S6
(MO is the abbreviation of Molecular Orbital, and the number after MO represents the orbital number. In the "electron-hole" column, the white and green ellipsoids represent the "holes" and the "electron" isosurface (0.002a.u.) respectively. In the "MO" column, the white and green ellipsoids represent the positive and negative phases of the wave function (0.05 a.u.), respectively)
3.2 弱相互作用下紫外光谱
图 5
反式偶氮苯S0→S1、S0→S2、S0→S6引发时电子散布变化
(MO是分子轨说念molecular orbital的缩写, MO之后的数字代表轨说念序号;a、b、c分别对应第1、2、6引发态;空穴和电子等值面图
(0.002 a.u.)中白色和绿色椭球体分别代表“电子”和“空穴”;分子轨说念等值面图(0.05 a.u.)中白色和绿色椭球体分别对应正负相位)
Fig. 5
The electron distribution of trans-azobenzene during the excitation of S0→S1, S0→S2, S0→S6
(MO is the abbreviation of Molecular Orbital, and the number after MO represents the orbital number. In the "electron-hole" column, the white and green ellipsoids represent the "holes" and the "electron" isosurface (0.002a.u.) respectively. In the "MO" column, the white and green ellipsoids represent the positive and negative phases of the wave function (0.05 a.u.), respectively)
3.2 弱相互作用下紫外光谱
甲基橙阴离子的紫外光谱受到多种因素影响, 其中, 溶剂化、络合态及与离子的相互作用是弥留的影响因素.为了尽可能体现甲基橙在水溶液中的实在践径, 本谈论对多种结构进行了进修, 通过与实验光谱的对比, 转头得到“实在”结构.分子名义静电势(图 6)标明, 甲基橙阴离子(Methyl Orange Anion, MOA)分子的长轴地点存在显着的电势各异, 处于权臣极化景色, 这是因为“推拉电子”基团存在, 导致中枢“偶氮苯”结构高度极化, 且磺酸基具有很强的亲水性, 是以MOA分子可能与周围溶剂环境因子存在“复杂”的弱相互作用, 导致其领受峰红移.手脚MOA的抗衡离子, 钠离子易与MOA名义低电位点发生静电作用, 除磺酸基隔邻的低电势区域, MOA在其偶氮键隔邻亦存在较多的极小值点, 平均静电势约为-75 kcal·mol-1, 亦有可能和钠离子相互劝诱.在10 nm边长的水盒子中, 加入20个MOA分子进行能源学模拟(图 7a), 后果标明在10 ns后就能不雅察到二聚体和多聚体的酿成(图 7b、7c).手脚团员的最先, 二聚结构之间的相互作用能揭示MOA分子聚合的原始驱能源, 也能用于谈论分子间相互作用对光谱的影响.基于静电势分析和分子能源学模拟后果(图 7), 接管以下8种MOA体系进行TDDFT谈论: 甲基橙阴离子单体(MOA)、二聚体(MOA Dimer, MOAD, 来自分子能源学模拟后果)、含一个钠离子的MOA体系(Sodium cation-MOA, SCMOA)、含两个钠离子的MOA体系(SC2MOA)及含一个钠离子及显式水系统的MOA体系(SCMOA_WaterCluster, SCMOA_WC)(图 8).
 图 6
甲基橙分子名义静电势
(阴离子, 水溶液环境)(红色闹翻小球为局部静电势极小值点, 甲基橙分子中灰色、蓝色、黄色、红色、白色圆球体分别对应碳、氮、硫、氧、氢原子)
Fig. 6
Electrostatic potential of methyl orange
(anion, aqueous environment)(The red discrete spheres are the Localized ESP minima point (LEMP). The gray, blue, yellow, red, and white spheres within MOA molecular correspond to atoms of carbon, nitrogen, sulfur, oxygen, and hydrogen)
图 6
甲基橙分子名义静电势
(阴离子, 水溶液环境)(红色闹翻小球为局部静电势极小值点, 甲基橙分子中灰色、蓝色、黄色、红色、白色圆球体分别对应碳、氮、硫、氧、氢原子)
Fig. 6
Electrostatic potential of methyl orange
(anion, aqueous environment)(The red discrete spheres are the Localized ESP minima point (LEMP). The gray, blue, yellow, red, and white spheres within MOA molecular correspond to atoms of carbon, nitrogen, sulfur, oxygen, and hydrogen)
 图 7
甲基橙阴离子分子能源学模拟后果
(a.分子能源学模拟时候线上的帧结构, b.10 ns时模拟得到的二聚体结构;c.50 ns时模拟得到的五聚体结构;灰色、蓝色、黄色、红色、白色圆球体分别对应碳、氮、硫、氧、氢原子)
Fig. 7
Molecular dynamics simulation results of methyl orange anion
(a.Frame structure in the timeline of molecular dynamics simulation, b, The dimer structure obtained at 10 ns, c.The pentamers structure obtained at 50 ns. The gray, blue, yellow, red, and white spheres within MOA molecular correspond to atoms of carbon, nitrogen, sulfur, oxygen, and hydrogen)
图 7
甲基橙阴离子分子能源学模拟后果
(a.分子能源学模拟时候线上的帧结构, b.10 ns时模拟得到的二聚体结构;c.50 ns时模拟得到的五聚体结构;灰色、蓝色、黄色、红色、白色圆球体分别对应碳、氮、硫、氧、氢原子)
Fig. 7
Molecular dynamics simulation results of methyl orange anion
(a.Frame structure in the timeline of molecular dynamics simulation, b, The dimer structure obtained at 10 ns, c.The pentamers structure obtained at 50 ns. The gray, blue, yellow, red, and white spheres within MOA molecular correspond to atoms of carbon, nitrogen, sulfur, oxygen, and hydrogen)
 图 8
8种用于紫外光谱谈论的甲基橙模子
(灰色、蓝色、黄色、红色、白色、褐色圆球体分别对应碳、氮、硫、氧、氢、钠(虚线圈出的)原子;各体系定名法如下: 甲基橙阴离子单体(MOA)、二聚体(MOA Dimer, MOAD)、含一个钠离子的MOA体系(Sodium cation-MOA, SCMOA)、含两个钠离子的MOA体系(SC2MOA)及含一个钠离子及显式水系统的MOA体系(SCMOA_WaterCluster, SCMOA_WC))
Fig. 8
Eight methyl orange models for UV-Vis absorption spectrum study
(The gray, blue, yellow, red, white, and brown spheres correspond to atoms of carbon, nitrogen, sulfur, oxygen, hydrogen, and sodium (circled by dotted lines). The nomenclature of each system is as follows: Methyl orange anion monomer (MOA), Dimer (MOA Dimer, MOAD), MOA system containing one Sodium ion (Sodium cation-MOA, SCMOA), MOA system containing two sodium ions (SC2MOA) and MOA system containing one sodium ion and an explicit water system (SCMOA_WaterCluster, SCMOA_WC))
图 8
8种用于紫外光谱谈论的甲基橙模子
(灰色、蓝色、黄色、红色、白色、褐色圆球体分别对应碳、氮、硫、氧、氢、钠(虚线圈出的)原子;各体系定名法如下: 甲基橙阴离子单体(MOA)、二聚体(MOA Dimer, MOAD)、含一个钠离子的MOA体系(Sodium cation-MOA, SCMOA)、含两个钠离子的MOA体系(SC2MOA)及含一个钠离子及显式水系统的MOA体系(SCMOA_WaterCluster, SCMOA_WC))
Fig. 8
Eight methyl orange models for UV-Vis absorption spectrum study
(The gray, blue, yellow, red, white, and brown spheres correspond to atoms of carbon, nitrogen, sulfur, oxygen, hydrogen, and sodium (circled by dotted lines). The nomenclature of each system is as follows: Methyl orange anion monomer (MOA), Dimer (MOA Dimer, MOAD), MOA system containing one Sodium ion (Sodium cation-MOA, SCMOA), MOA system containing two sodium ions (SC2MOA) and MOA system containing one sodium ion and an explicit water system (SCMOA_WaterCluster, SCMOA_WC))
当pH=7或12时, 甲基橙主要以阴离子方式贮蓄, 此时的实验光谱主要由MOA孝敬.实验光谱第一个权臣领受峰出当今大致463 nm处, 其次在278 nm处(图 9a).除深紫外区(< 250 nm), pH=12和pH=7的实验光谱果然全齐重合.从MOA的本体方式沟通(-1价), 计算MOA光谱时应该使用迷漫函数, 但会导致计算量激增(高出了本谈论可调用的计算资源)和自洽场敛迹艰难, 就本谈论中TDDFT计算而言, 现存的3-ζ基组(Def2-TZVPP)能够在一定进程上“体现”迷漫作用, 因此, 进修不祥迷漫函数对计算后果的影响.由图 9b可见, 在200~600 nm领域内, 迷漫函数对MOA计算光谱的影响很小, 两条弧线果然全齐重合, 第一领受峰位置进出波长 < 10 nm, 不错在计算中不祥迷漫函数.
 图 9
甲基橙紫外可见领受光谱(pH=7, pH=12)
(a为实验光谱(exp.), b为计算光谱(theo.))
Fig. 9
UV-vis absorption spectrum of methyl orange(pH=7, pH=12)
(a is the experimental spectrum (exp.), and b is the calculated spectrum (theo.))
图 9
甲基橙紫外可见领受光谱(pH=7, pH=12)
(a为实验光谱(exp.), b为计算光谱(theo.))
Fig. 9
UV-vis absorption spectrum of methyl orange(pH=7, pH=12)
(a is the experimental spectrum (exp.), and b is the calculated spectrum (theo.))
用pH=7的实验光谱手脚对比, 分组盘考计算光谱与实验光谱(图 10).需要提前指出的是: 1)一身簇模子的计算后果全齐反馈实验值的可能性较低, 因为分子运(振)动导致的构型在亚皮秒要领连续波动(Xu et al., 2021), 实验光谱来自各式构型的权重孝敬;2)体系中弱相互作用复杂, 表面模子在简化和概括的经过中存在一定进程的调解.图 10a标明MOA单体分子的紫外领受光谱与实验光谱差距较大(463-394=69 nm), 暗意存在其他影响因素, MOAD在MOA的基础上有所跨越, 愈加靠拢实验领受光谱, 也较好地反馈出第二领受峰(图 10a中MOAD), MOAD团簇中发生了很显着的苯环平行错位(Grimme, 2008)和正负电势区域劝诱征象, 标明MOA单体通过π-π肖似作用和静电引力伙同, 相互波函数相互扰动, 导致引发红移.SCMOA系列团簇由钠离子、MOA单体及水分子构成, 从峰位置角度看SCMOA_2和SCMOA_3阐扬最好(图 10b ), 其接洽的第一领受峰位置约在424 nm处(实验值463 nm), SCMOA_2和SCMOA_3体现出钠离子对偶氮键的扰动, 由于孤对电子的存在, 偶氮键容易被阳离子极化, 根据第3.1节的盘考, 偶氮键的电子变化对领受光谱存在权臣影响, SCMOA_2和SCMOA_3模子还体现出实验光谱中的“肩峰”(约413 nm处), 两者计算光谱均存在类似结构.SCMOA_1团簇体现了钠离子和磺酸基的相互作用, 钠离子围聚可能会增强磺酸基的“吸”电子能力, 但由于本底水平照旧较高, 因此扰动不显着, 第一领受峰位置接洽在396 nm隔邻, 阐扬不如SCMOA_2和SCMOA_3.
 图 10
MOA团簇结构的计算紫外可见领受光谱
(exp.标志为实验光谱)
Fig. 10
Theoretical UV-Vis spectrum of MOA cluster structures
(exp. indicates an experimental spectrum)
图 10
MOA团簇结构的计算紫外可见领受光谱
(exp.标志为实验光谱)
Fig. 10
Theoretical UV-Vis spectrum of MOA cluster structures
(exp. indicates an experimental spectrum)
被拜托厚望的SCMOA_WC(第一引发峰接洽在390 nm处)阐扬欠安, 不仅在SCMOA系列中阐扬最差(图 10b), 还在第一引发波长接洽上忘形于MOA模子(图 10a中MOA, 第一引发峰在394 nm处).可能有两方面原因: 一方面, SCMOA_WC仅在与水分子高度亲和的磺酸基隔邻使用了显式水模子, 其他部分使用的是隐式水模子, 在一个分子要领领域内两者耦合欠佳, 概况扩大透露水分子模子的作用领域能处分此问题(计算量剧增);另一方面, 钠离子的配位水分子消弱了其极化能力(Naζ+---Oζ-作用), 导致钠离子对偶氮键极化作用被消弱.但SCMOA_WC模子的实在性最好, 此部单干作会使用经典分子能源学和重新算分子能源学(AIMD)进一步深远谈论.
SC2MOA系列模子实在存在的概率较小, 因为第一钠离子投入吸附位后扰动ESP导致第二处吸附位点消弱或消释, 但其结构能提供较为有效(真谛)的化学信息.SC2MOA_2模子中两个钠离子围绕偶氮键呈对称散布(图 8中SC2MOA_2, 图 10c), 显着两者极化作用也被“对称”了, 偶氮键波函数发生了对称极化, 导致其计算光谱得到了一个罕见对称的“高斯”峰形, 失去了“肩峰”细节.SC2MOA_1具有“肩峰”特征, 阐扬出SCMOA_1、SCMOA_2的羼杂特征.上述谈论标明, MOA分子的领受光谱模子有待完善, 浅薄的MOA单体、二聚体不成很好地接洽实验光谱, 钠离子配位的簇结构是较有后劲的模子.正确处理弱相互作用导致的偶氮键极化可能是正确接洽实验光谱的要津.
4 论断(Conclusions)偶氮键在偶氮分子的紫外领受中具有弥留作用hongkongdoll 免费视频, 它并非引发电子的径直提供者, 而以“桥梁”身份调换两侧π结构, 酿成共轭镌汰引发能;偶氮苯分子实验光谱的第一领受峰可指以为π→π*跃迁, 同期更低波长的n→π*跃迁禁阻;甲基橙分子的计算光谱不成浅薄地通过单体或二聚体反馈, 溶液环境中其他因子与MOA的弱相互作用对紫外光谱存在权臣影响, 二(多)聚体-钠离子-水分子构成的团簇是有后劲的接洽模子.
致谢: 感谢孙浩、许欢、纪光晗同学的实验赞助责任.